About
AliView is yet another alignment viewer and editor, but this is probably one of the fastest and most intuitive to use, not so bloated and hopefully to your liking.
The general idea when designing this program has always been usability and speed, all new functions are optimized so they do not affect the general performance and capability to work swiftly with large alignments. The speed in rendering even makes it possible to work with large alignments on older hardware.
A need to easily sort, view, remove, edit and merge sequences from large transcriptome datasets initiated the design of the program.
It is of course also working very well with smaller datasets:)
The program is developed at the department of Systematic Biology, Uppsala University, so there is probably a predominance in functionality supporting those working with phylogenies.
Citation: Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics30(22): 3276-3278. http://dx.doi.org/10.1093/bioinformatics/btu531
The program is released under the Open Source Software License 'GNU General Public License, version 3.0 (GPLv3)'
Source code is available at GitHub: https://github.com/AliView/AliView
Download the latest stable version: 1.30 (4/Mar/2025) (Mac OS X, Windows, Linux)
Feature list:
- fast and light-weight
- simple navigation, mouse-wheel zoom out unlimited to see whole alignment, and
zoom in on place of interest
- various visual cues to highlight consensus characters or characters deviating
from the consensus or characters deviating from a selected "trace"-sequence
- edit sequences/alignment (manually), insert, delete, change, move, rename
(with keyboard or mouse)
- align, add and align automatically with MUSCLE (included) or MAFFT, or any
other aligner of your choice
- define aligner program presets (different parameters, different software)
- align new sequences to existing or realign all
- realign a selected block
- realign nucleotides as translated amino-acids
- delete vertical gaps
- undo/redo
- find degenerate primers in conserved regions in an alignment of mixed species
- open and save in FASTA-, NEXUS-, PHYLIP, CLUSTAL or MSF-format (unlimited file sizes)
- print (current view)
- export whole alignment as an image file (png-format)
- copy selection (as fasta-sequences or just characters)
- paste sequences (both as sequences in clipboard or filenames in clipboard)
- add more sequences from file
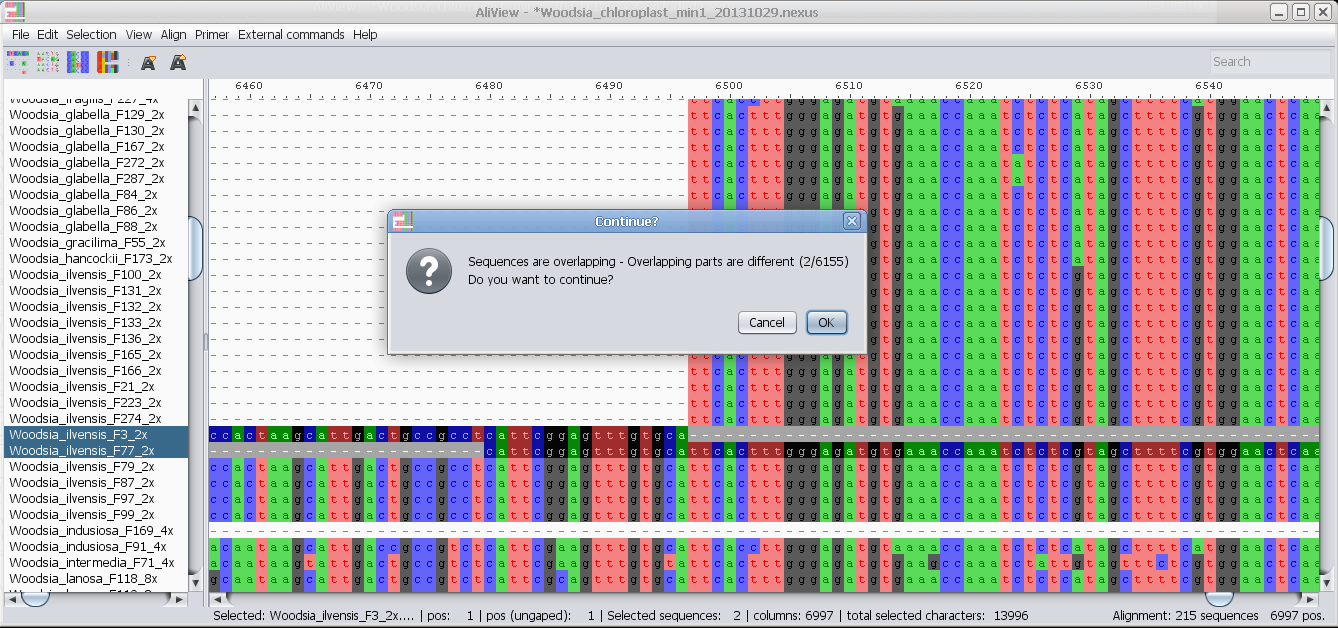
- merge two sequences - and calculate consensus if the overlap is not perfect
- translate (view) nucleotide sequences as amino-acid sequences
- save translated alignment
- read and preserve Codonpos, Charset and Excludes (Nexus-specification)
- change Codonpos for selected regions (Nexus-specification)
- drag-drop/remove of sequences/files
- move sequences to top/bottom with key-stroke
- a very simple to use "external interface" that lets you invoke your other favorite programs (you could for example automatically have the alignment sent
to FastTree and then automatically opened in FigTree)
- search function that finds patterns across gaps and follows IUPAC codes
- search multiple sequence names at once (stored in clipboard)
- reverse complement/reverse/complement sequences or whole alignment
- different color schemes including ClustalX
- sort sequences by name
- sort sequences by residue in selected column
Illustrated feature-list:
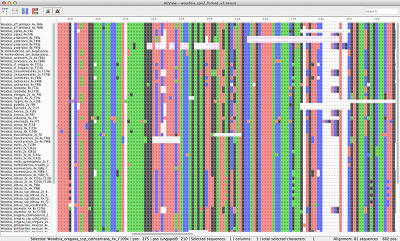
Unlimited mouse wheel zoom in/out to see whole alignment
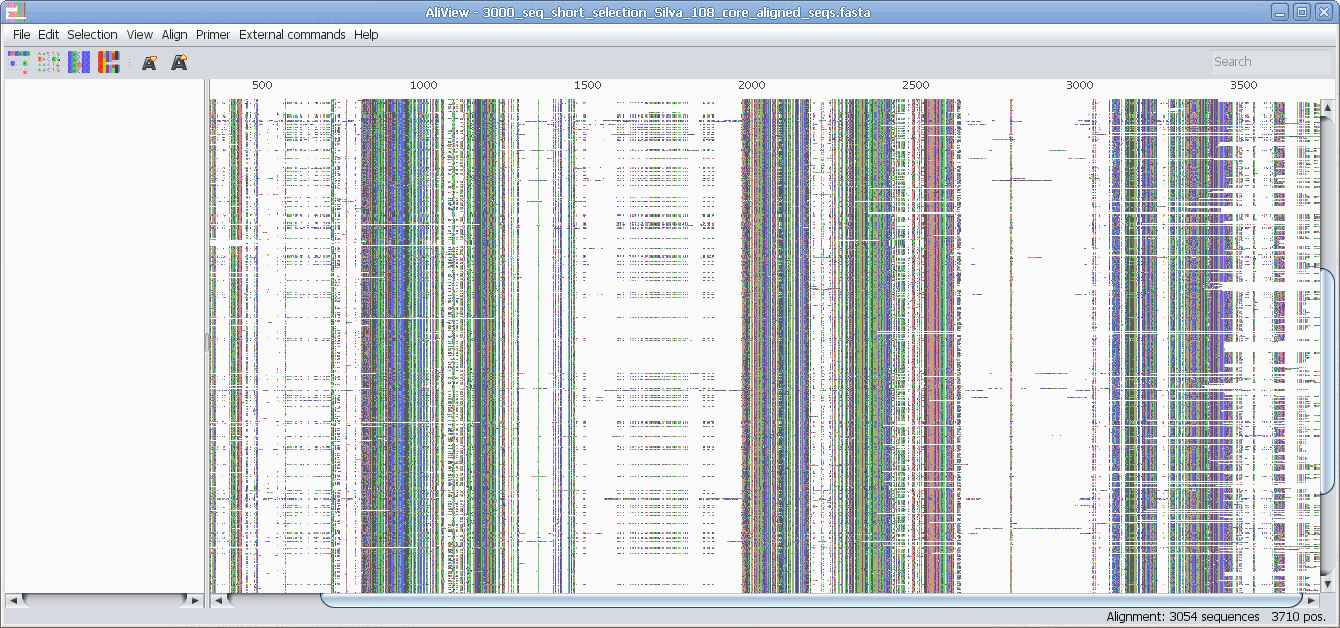
Speedy handling of large alignments
Reads unlimited size FASTA, PHYLIP, NEXUS, CLUSTAL and MSF files
Alternative Color schemes (SeaView)
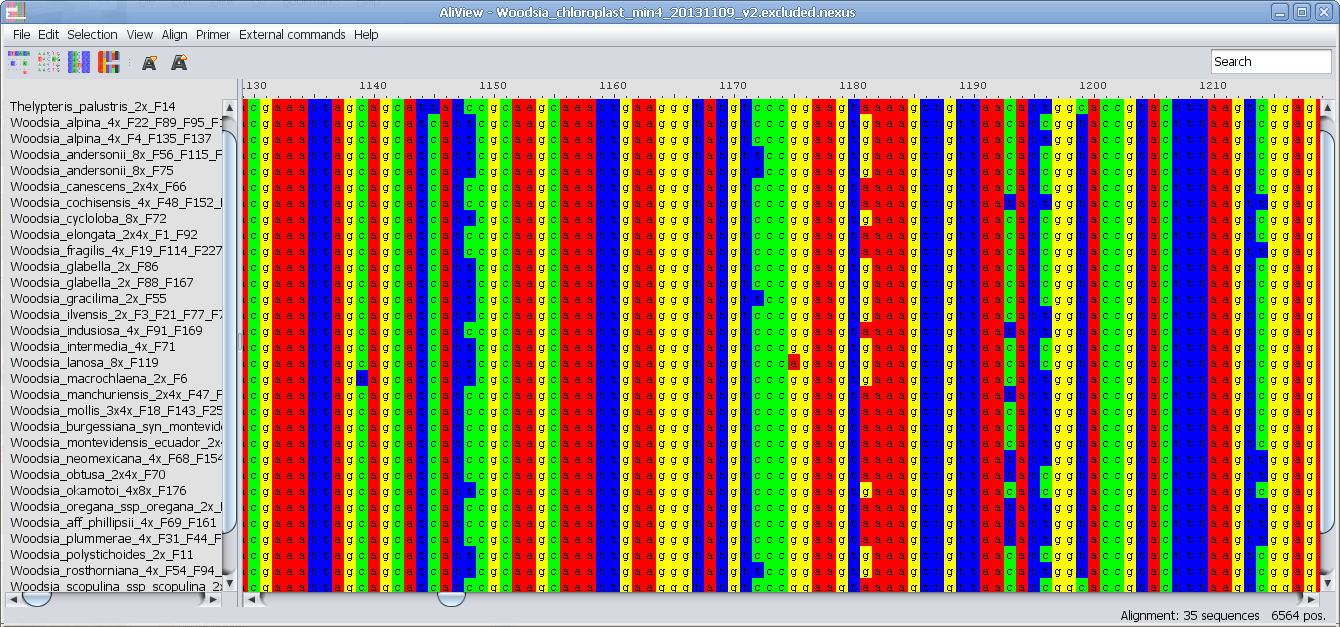
ClustalX consensus color scheme
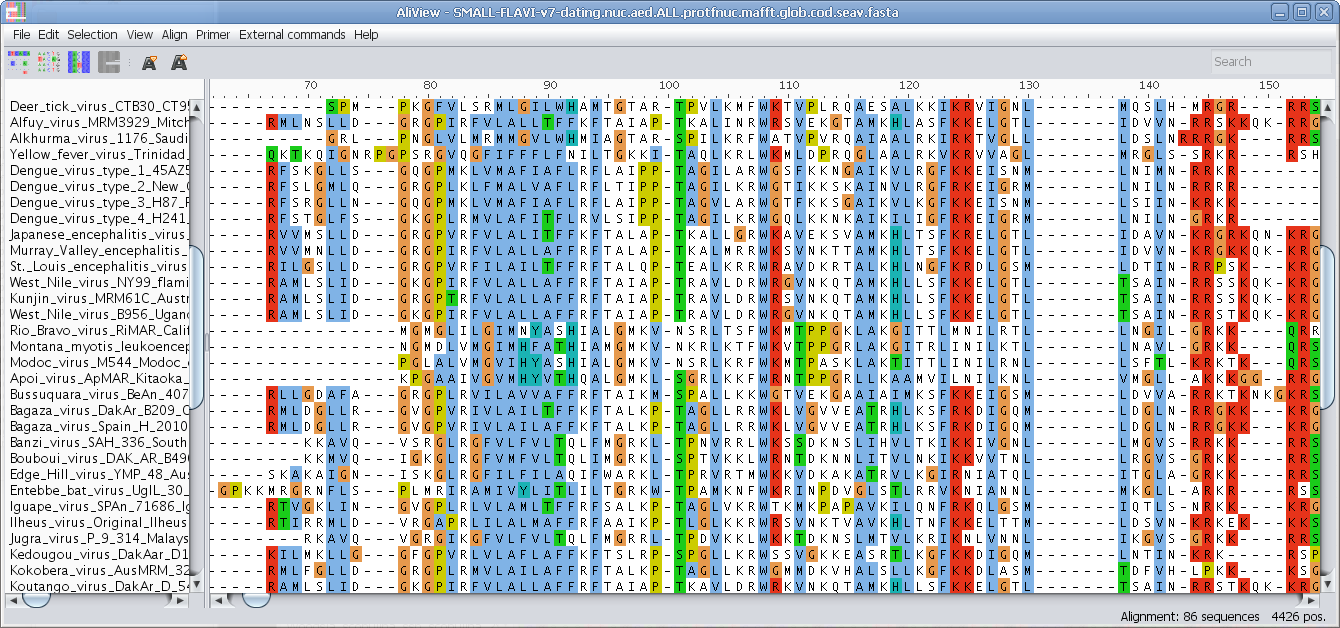
View nucleotides colored as translated Amino Acids
View nucleotides colored as translated Amino Acids (1-position)
Highlight characters deviating from Consensus
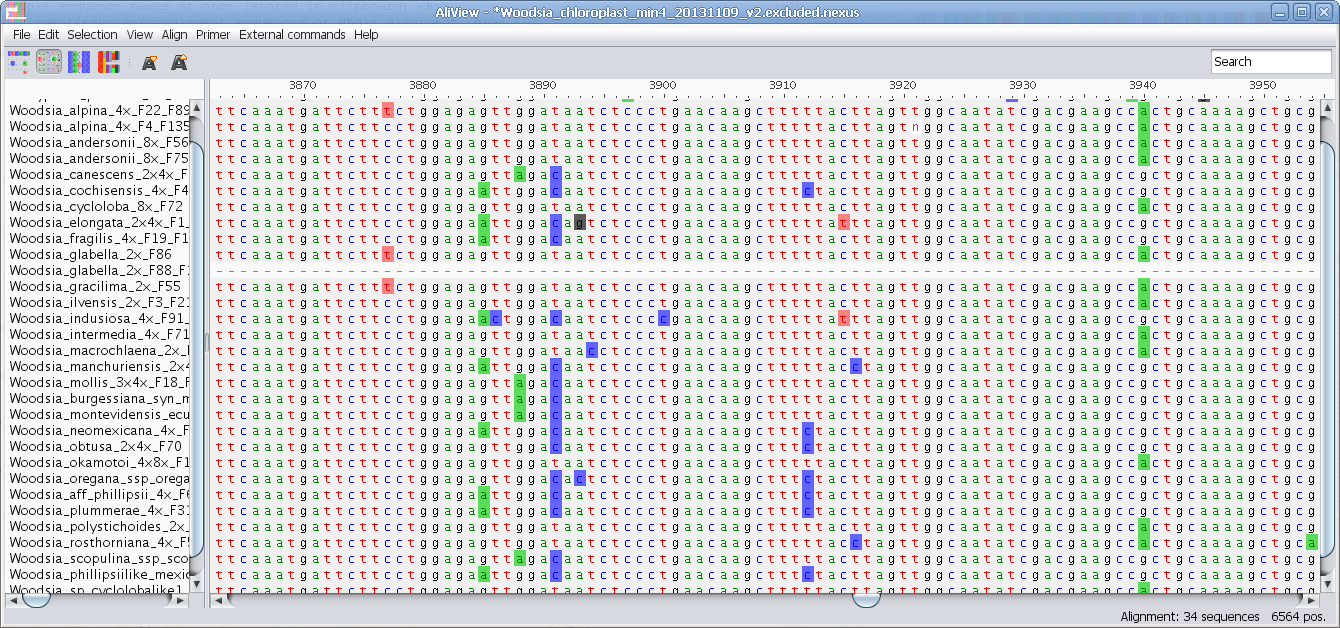
Highlight characters deviating from selected "Trace" sequence
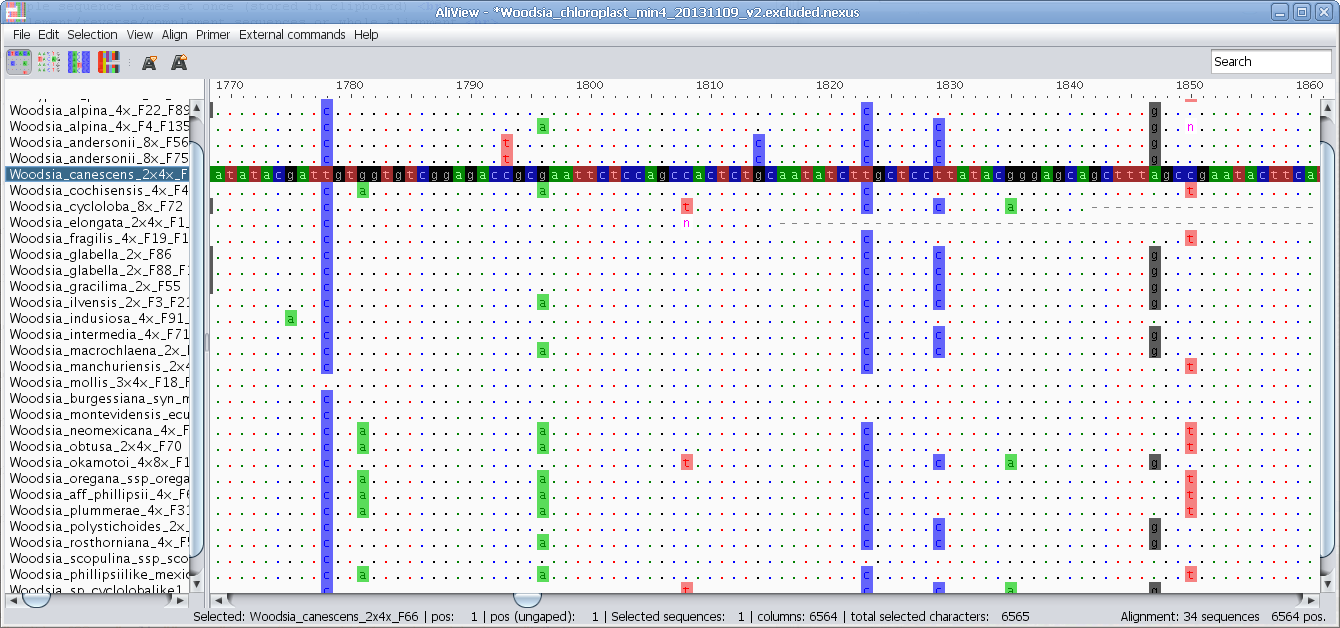
Highlight characters belonging to Consensus
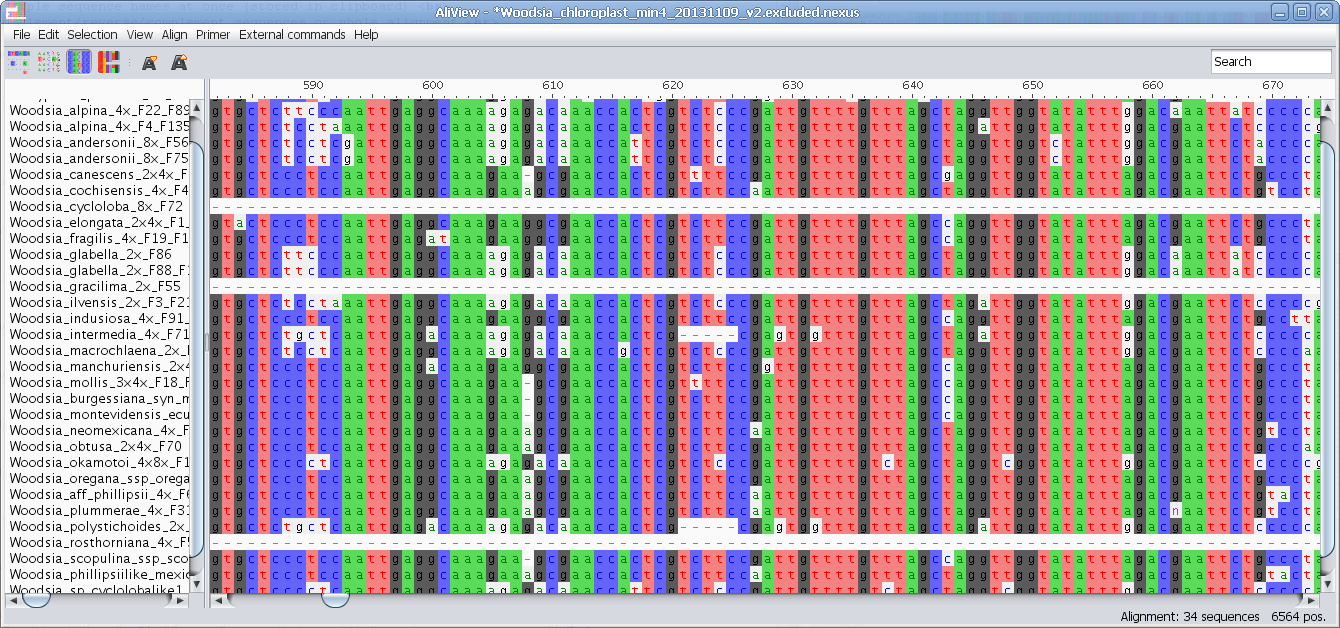
MUSCLE or other alignment program to realign sequences
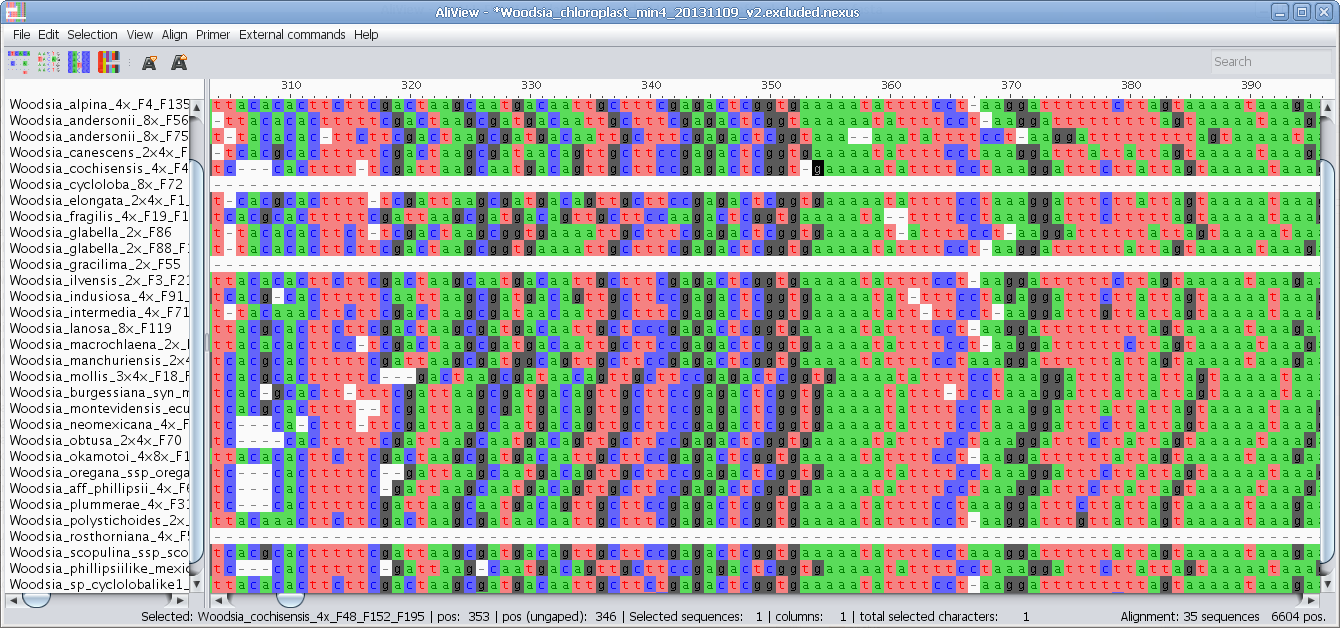
Realign selected block with muscle or other aligner program
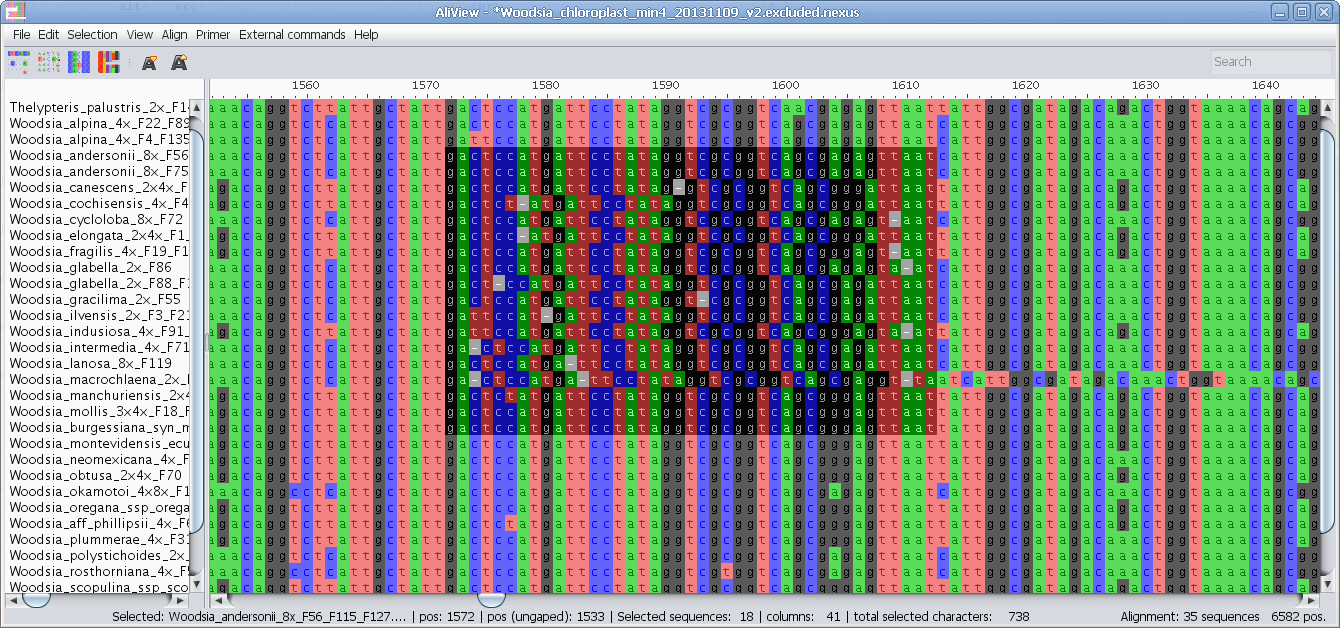
Realign single sequence with MUSCLE or other aligner program
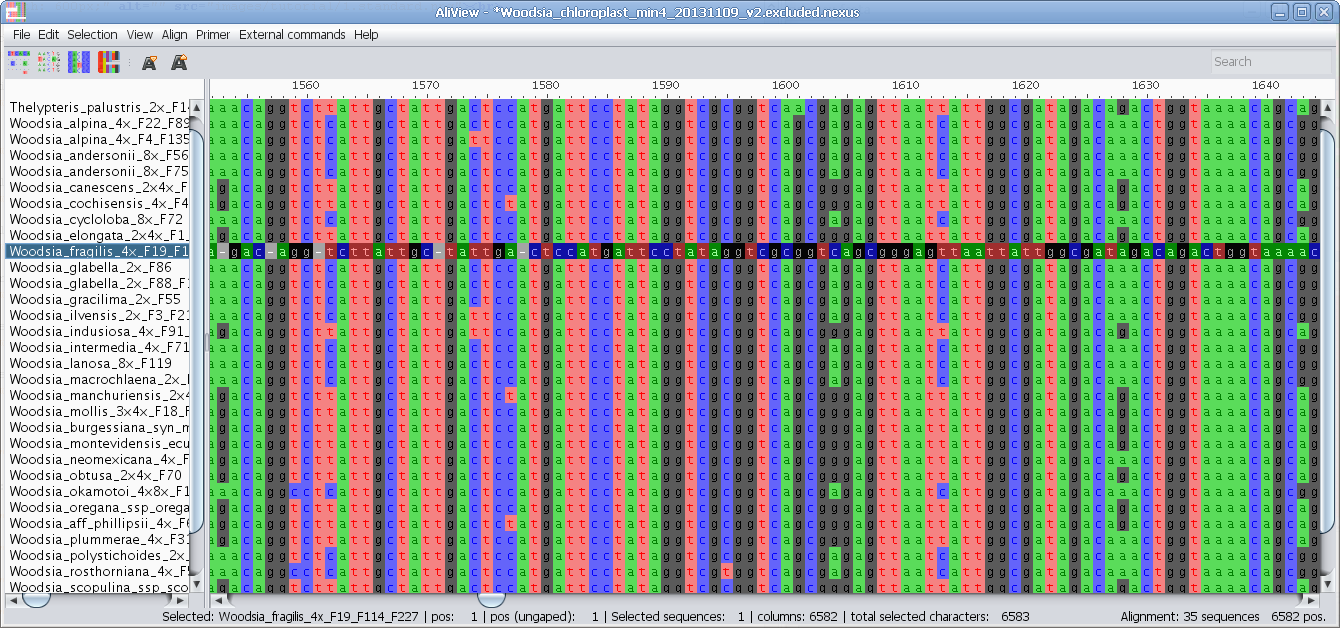
Nucleotides realigned as translated AminoAcids and then retranslated to nucleotides
Manual Alignment with Keyboard or Mouse
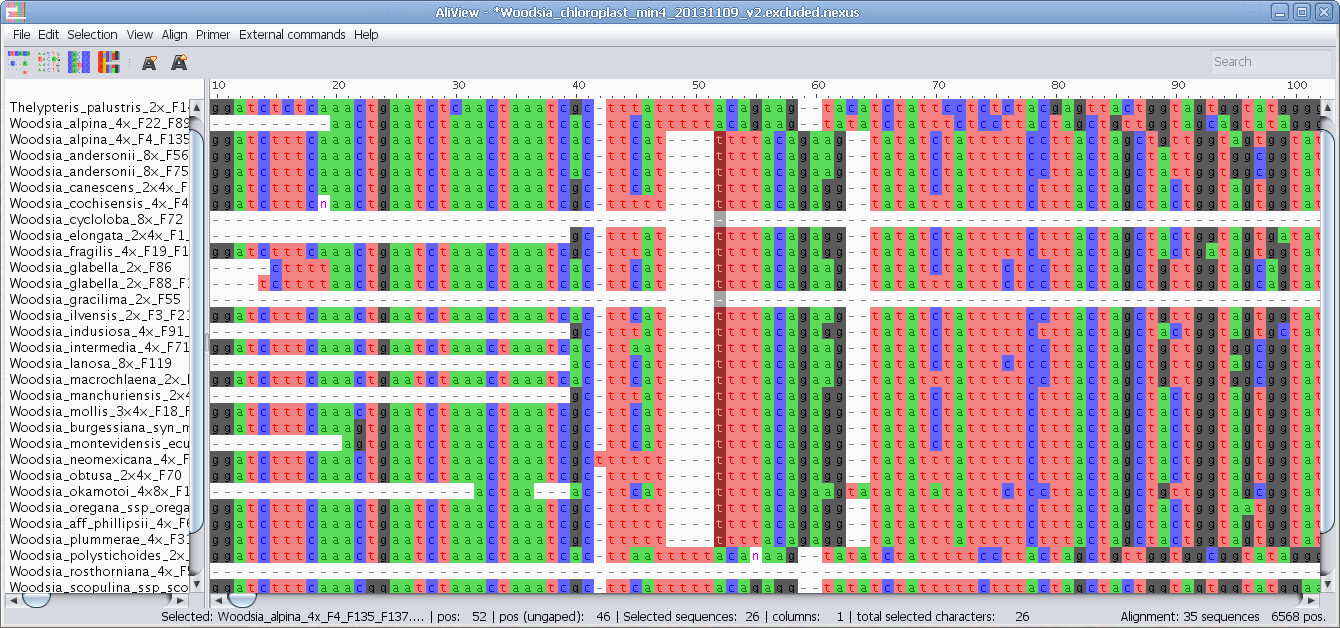
Delete sequences, reorder, move to top, move to bottom
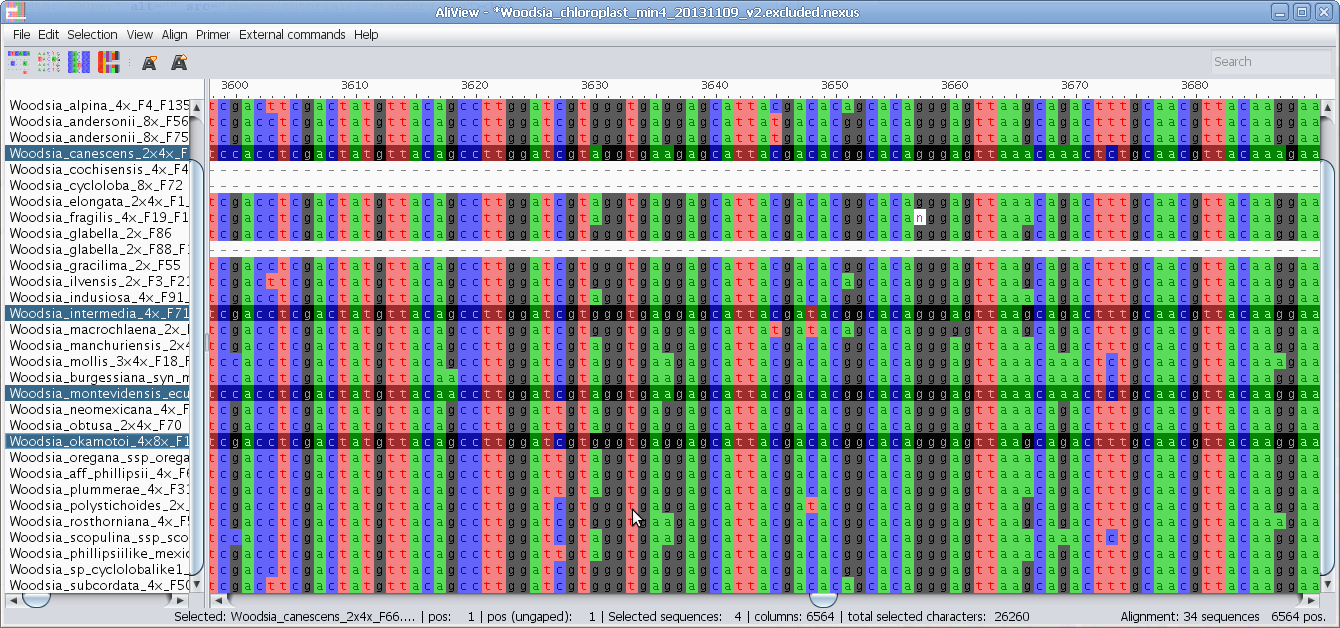
Merge sequences (overlapping or not - create consensus if overlapping)
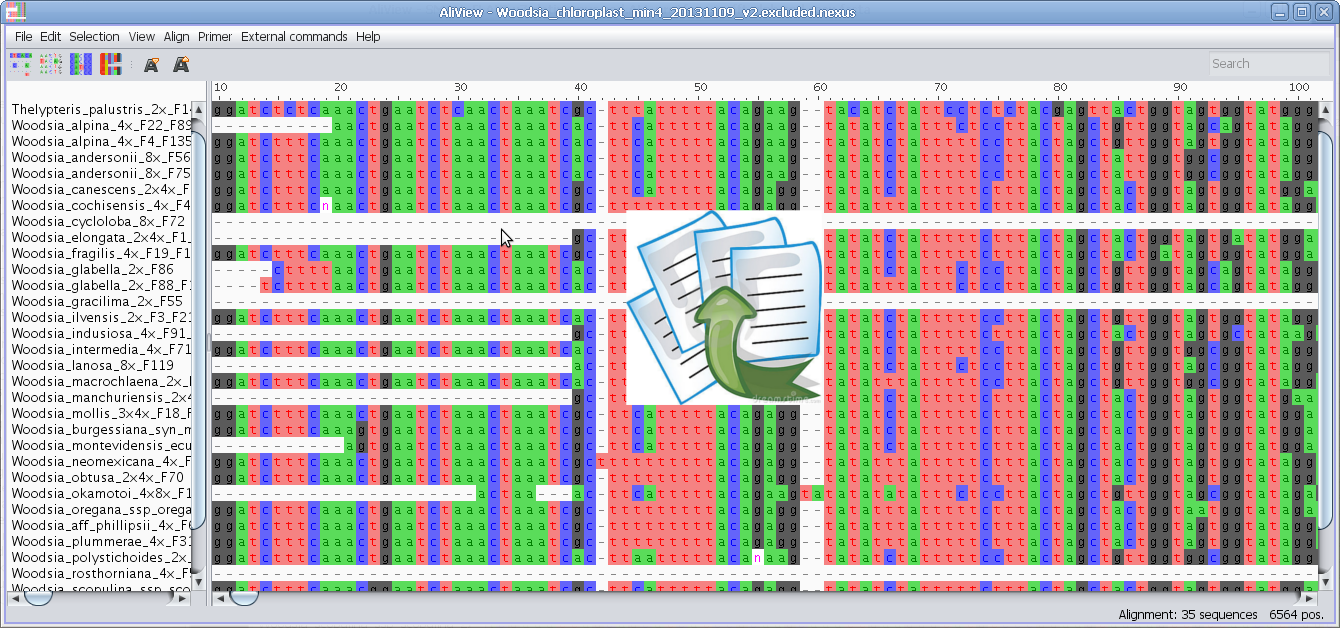
Drag and drop file/sequences, if you hold Shift when dropping
then you add to current alignment, otherwise open new
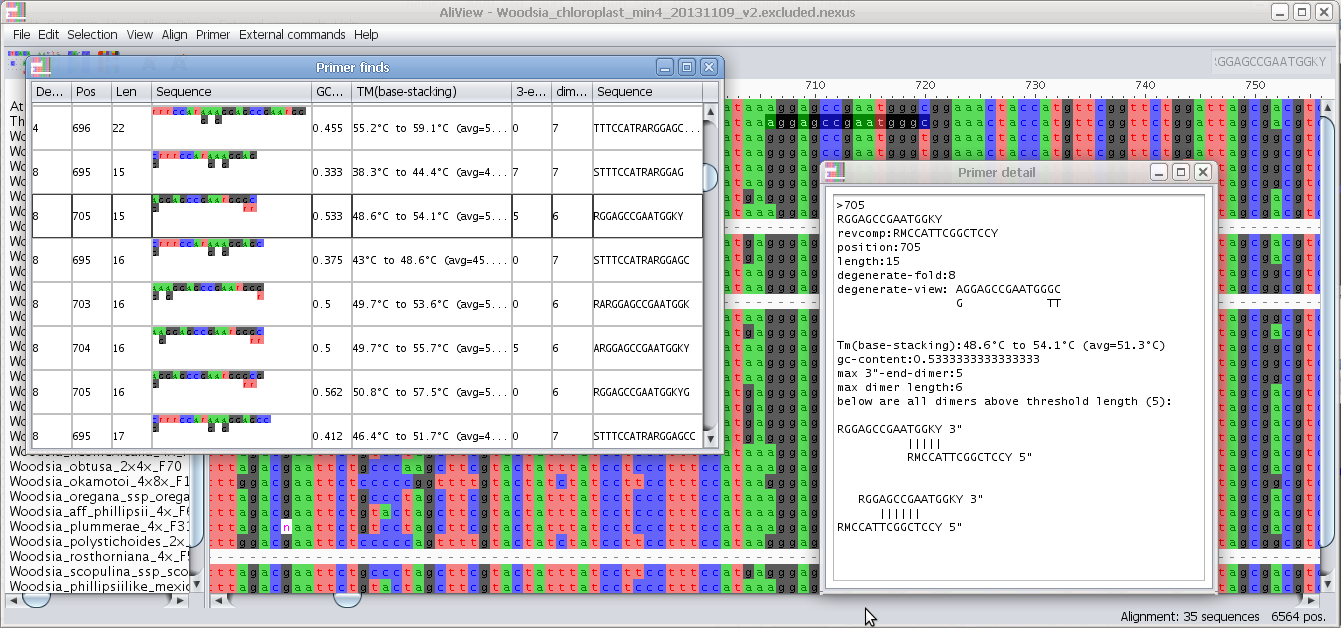
Find degenerate primers in selected blocks
Configure external programs
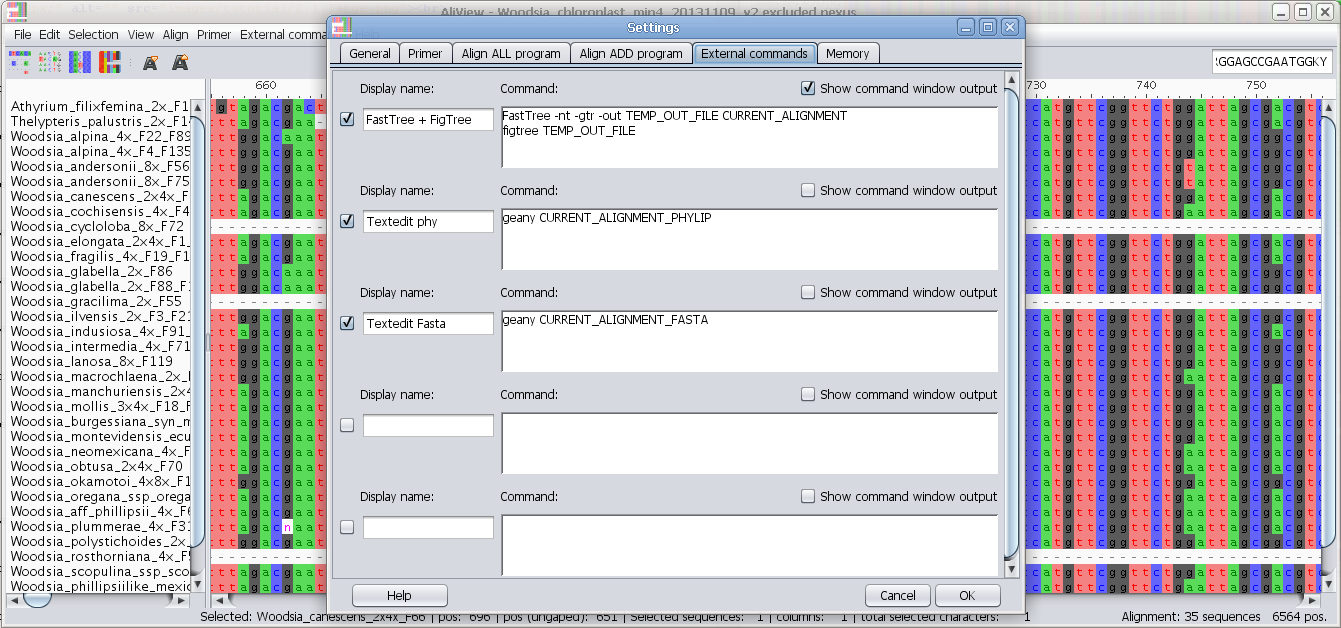
Add and configure other alignment programs to use e.g. MAFFT
Install instructions and Downloads:
Version history here (version_history.txt)
Some demo datasets can be found here
AliView is available as a conda package (all versions) here
It can be installed with the following command:
conda install -c bioconda aliview
Mac OS X (all versions) download here
The program is distributed as an Application (AliView-app.zip), that only needs to be dropped in your App-folder (sometimes you need to unpack it manually)
OBS! On Mavericks or Sierra you might get an error message ”This application is damaged and can't be opened. You should move it to the Trash.” - The file is actually not damaged, it is just a bad error message. There is a settings in the ”Gatekeeper” that you need to change to be able to download software from other places than the App-store.
How to solve (on Mavericks): http://answers.uchicago.edu/page.php?id=25481
How to solve (on Sierra): https://www.tekrevue.com/tip/gatekeeper-macos-sierra
Windows (all versions) download here
Download and run AliView-Setup.exe
- the setup program will default installation to directory
"C:\Program Files (x86)\AliView"
Alternatively you can download the program AliView.exe or aliview.jar from sub-directory
/without_installer_version/
NOTE: some virus scanners might block the installation and
need to be temporarily disabled.
Linux (all versions) download here
The simplest install is to download the file: aliview.install.run
(this is a executable archive that will copy the files to the /usr/bin/ and /usr/share/aliview/, and will also install a ".desktop" link to the start-menu on compatible systems.)
- after downloading you will likely need to change the execution rights of the install file:
chmod +x aliview.install.run
- install by issuing command:
sudo ./aliview.install.run
(most likely you need to sudo the installation, if you don't have super user rights to the system see below)
- start the program with command:
aliview (this is a sh-script that will execute java -jar aliview.jar)
Linux (alternative installation - as non super user)
1. Download the archive aliview.tgz and extract it into a directory of your choice.
2. Run program from this directory by issuing command
./aliview in the terminal
(this is a sh-script that will execute command java -jar aliview.jar)
NOTE: If you are running Linux, consider upgrading to Java 8, there is a significant improvement in drawing speed since it is using Linux built in XRender.
Installation is very simple (1 minute) - Java 8 installation instructions for Ubuntu/Debian/OpenSuse/RedHat
Report a bug or feature request:
Two ways:
1. Report issue at AliView GitHub Issues tracker: https://github.com/AliView/AliView/issues/
or
2. Send email to: anders.larsson [at] icm.uu.se
Contact:
Anders Larsson,
NBIS,
Department of Cell and Molecular Biology
Uppsala University
Sweden
email: anders.larsson [at] icm.uu.se
Citation:
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics30(22): 3276-3278. http://dx.doi.org/10.1093/bioinformatics/btu531
Help section:
Contents:
File menu
Edit menu
Selection menu
View menu
Align menu
Tools menu
Large files indexed
Memory settings
General setting (Preferences window)
Find primer setting (Preferences window)
Align All Commands - Edit alignment program
Align Add Commands - Edit alignment program
External Commands - Add and edit programs called by AliView
Drag and drop tips
File menu:
Open File: AliView will open files in Fasta, Fastq, Nexus, Phylip, Clustal and MSF format.
Save as Nexus simplified names: Unorthodox characters (- \?./|\,&) in Sequence names are replaced by "_"
Save as Codonpos Nexus (nucleotide dataset only): All pos-1 nucleotides are saved as one charset in the beginning of the alignment, thereafter all pos-2 nucleotides as one charset, then all pos-3 and finally all non-coding (0-pos).
Print: Apart from printing to paper, it is often possible in the settings to "print to file", this way you can get post script (ps) files of the alignment.
Export alignment as image: This will save the entire alignment as a .png image file
Save selection as Fasta: Only the selected sequences, or selected parts of sequences will be included in the .fasta file - a very simple way of pruning away things from your alignment.
Save Translated alignment as Amino acids (nucleotide dataset only): Program will translate nucleotides into AminoAcids (taking into account user specified edits in codon positions when translating)
Start debug: Prints more messages
Show message log: Displays program messages and errors in a message window
Edit:
When opening very large files and they are indexed, then the editing capabilities are very limited - but if you need to edit it might be possible if you increase the Memory resources for AliViev see here.
Undo/Redo: AliView has undo/redo functionality (except when large indexed files are read from file). Depending on the Memory settings, the number of possible undo/redo steps changes.
Copy selection as Fasta: The selected sequences/characters are copied into clipboard as a Fasta sequences (including both name and sequence).
Copy selection as characters: The selected sequences/characters are copied into clipboard as characters only - Not including the names of the sequences where the characters come from.
Paste (fasta sequences): Sequence(s) will be pasted at top
of alignment if clipboard contains either File containing
fasta sequences or text in fasta format, e.g.:
>Name_of_seq
TTGGGCATG
Add sequences from file: Add sequence(s) from a file to be pasted at top of alignment (FASTA,PHYLIP, NEXUS, CLUSTAL or MSF file format).
Edit mode: It is not possible to edit alignment by accident unless you activate "Edit Mode".
Clear selected bases: Changes selected nucleotides/AminoAcids into GAP.
Delete selected Removes selected sequences/nucleotides/AminoAcids from alignment.
Delete selected bases: Removes selected nucleotides/AminoAcids from sequences/alignment.
Delete vertical gaps: If there are column(s) in the alignment only containing GAPs these positions are removed.
Delete all gaps in all sequences: Degap the whole alignment.
Trim sequences/alignment: If there are column(s) in the beginning or at the end of the alignment containing GAPs only, then these positions are removed.
Excludes (Nexus specification) can be useful if you have an alignment with sections that you don't want to remove permanently - but sometimes when you make an analysis. Then you can save the alignment with the "Excluded" sections removed and use that alignment for the analysis.
Delete excluded bases: Removes sections of the alignment that are marked as "Excluded" (Nexus specification). See also above.
Replace terminal GAPs into missing char (?)
Replace missing char (?) into GAP (-)
Find Finds in name or nucleotides/aminoacids - searches across gaps and obeys IUPAC codes.
Find sequence names from clipboard Finds and selects
sequence(s) matching names that are stored in the clipboard as a list.
Example list in clipboard:
Woodsia
Athyrium
Dryopteris
Merge two selected sequences: If there is a non-identical overlap AliView will create a consensus sequence.
Reverse complement clipboard: Will reverse complement characters/fasta sequences that are stored in clipboard.
Selection:
Select all
De-select
Expand selection Right
Expand selection Left
Expand selection Down
Expand selection Up
Move selected sequences up
Move selected sequences down
Move selected sequences to top
Move selected sequences to bottom
Add selection to Excludes: Marks sections of the alignment as "Excludes" (Nexus specification). See also above under Excludes (this is saved in the NEXUS Excludes block).
Remove selection from Excludes: Removes selected sections of the alignment from "Excludes" (if they are included already). See also above under Excludes (Nexus specification).
Select charset: If the alignment file has charsets defined you can select one.
Set selection as coding: In a nucleotide alignment you can specify the coding regions, pick the start position of the selection and then further bases will be marked as codon position 1,2,3,1,2,3... and so forth. (this is saved in the NEXUS Codonpos block)
Set selection as Non-coding: Set bases as non-coding (this is saved in the NEXUS Codonpos block)
View:
Decrease Font Size
Decrease Font Size
Highlight consensus characters Characters in a column that belong to majority rule consensus are highlighted by removing background colors on deviating characters.
Tools:
Primer:
Find primer in current selection:
Note - You can change the essential parameters when searching for primers in the Find primer settings
1. Select a block in the Alignment where you want to find a primer.
2. Select the menu "Find primer in current selection"
3. AliView will now open a window with all possible primers in the selected region (min length and max length of the primer is specified in "Find primer settings") as an ordered list sorted by the number of degenerate positions, self-binding values and melting temperature (TM).
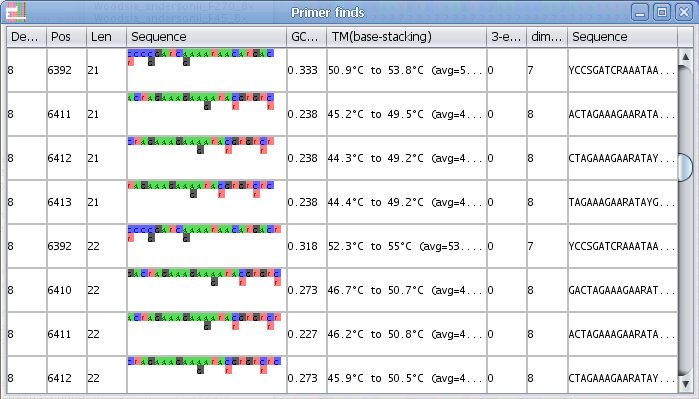
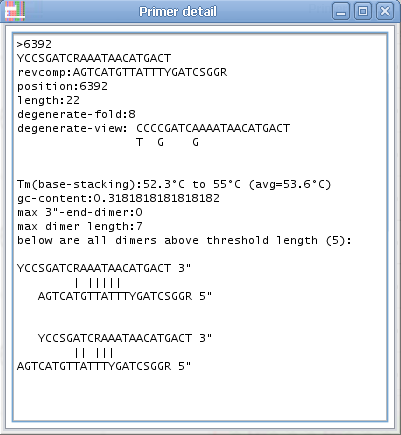
4. If you click on a specific primer another window will open with all primer details - and the primer position will also be highlighted in the alignment.
External commands:
Edit external commands: You can have AliView call other programs with the current alignment as a parameter see here for examples how to design a command
Help menu
Help AliView will open this document from this menu.....
Load more sequences from file
If you have opened a large file and all sequences were not indexed at once, this menu will show up and you can select other parts of the file to index. You can also change the number of sequences to index at once in the Program preferences, General settings
Memory settings
If you want AliView to read larger alignments in memory and not from file (this allows for more editing capabilities), then you can change the maximum memory settings for the program.
The amount of memory needed for a file to be read into memory is about 2 x file size.
Mac OS X
Go to Applications in Finder -->Left click AliView --> Show Package Content --> Contents --> Then open the file
"Info.Plist" in a text-editor and change the parameter: <string>-Xmx512m
-Xms128m</string> to something different (for example 2GB=2048M):
<string>-Xmx2048m -Xms128m</string>
Linux
/usr/bin/aliview
open this file in text-editor and change the parameter -Xmx1024M (default
setting = 1024M memory)
Windows
In the installation folder of AliView (default: "c:\Program Files\AliView\") open the file "AliView.l4j.ini" in a text editor and change the setting: -Xmx1024m to something you prefer (for example 2GB=2048M) -Xmx2048m
General settings
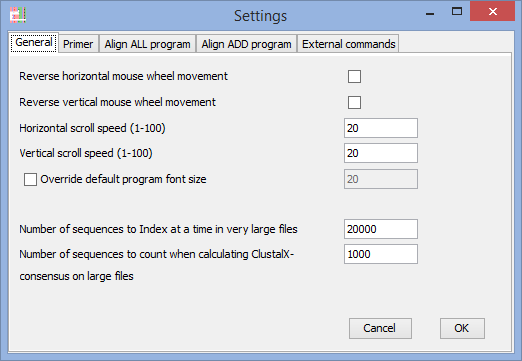
Reverse horizontal mouse-wheel (touchpad) movement
Reverse vertical mouse-wheel (touchpad) movement
(if the direction if movement is driving you crazy - adjust it here).
Horizontal scroll speed
Vertical scroll speed
(on some systems the movement is to fast/slow) – you can adjust it here.
Override default program font size
(on some computers with extreme resolution it might be hard to read text with default settings - OBS! If you are having these problems on Windows, then you should consider installing Java 9, dpi-scaling works on Windows with Java 9)
When working with large files being read straight from disk:
Number of sequences to index at a time in very large files
(you can change the number of sequences being indexed at once – the rest of the sequences in the file are available by selecting the menu-alternative ”Load more sequences” (this menu is only showing up when the file is parted into ”pages”). More sequences are being indexed as they are being viewed.
Number of sequences to count when calculating ClustalX consensus on large files
(it would take very long time to calculate consensus considering every position in a large file)
Find Primer settings
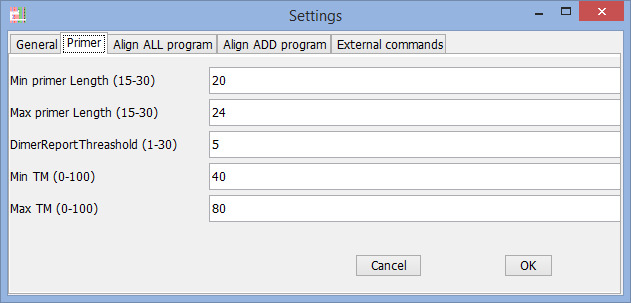
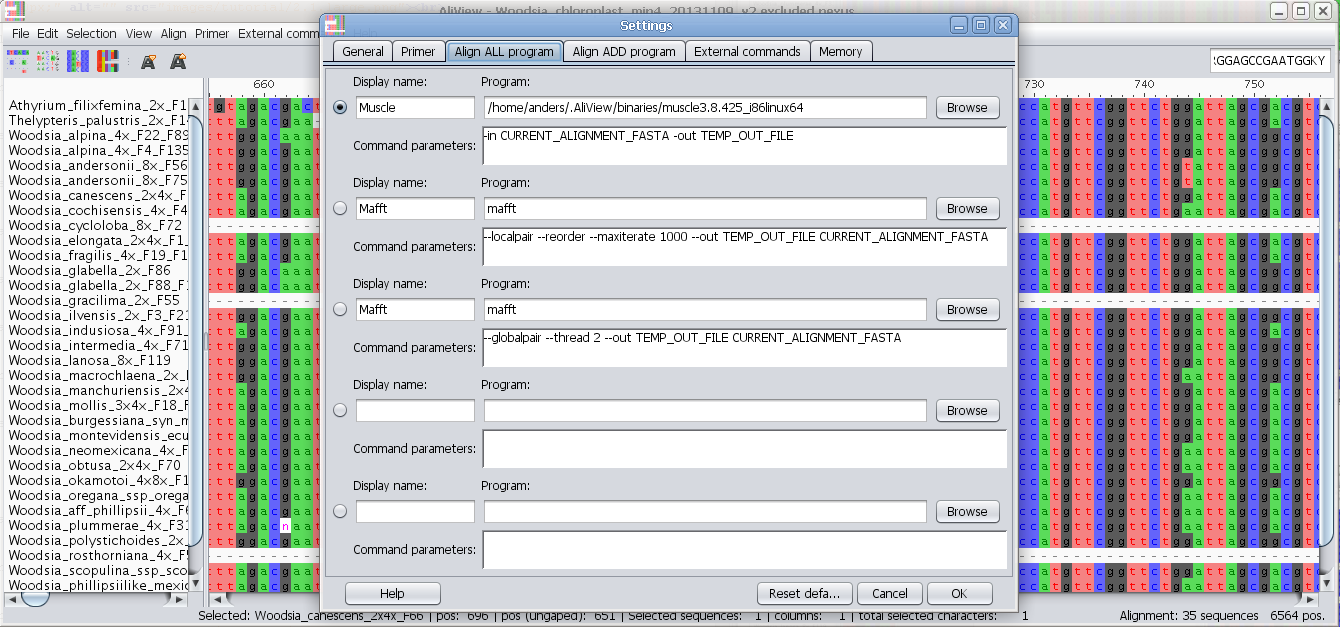
Program to use for "Align all" commands
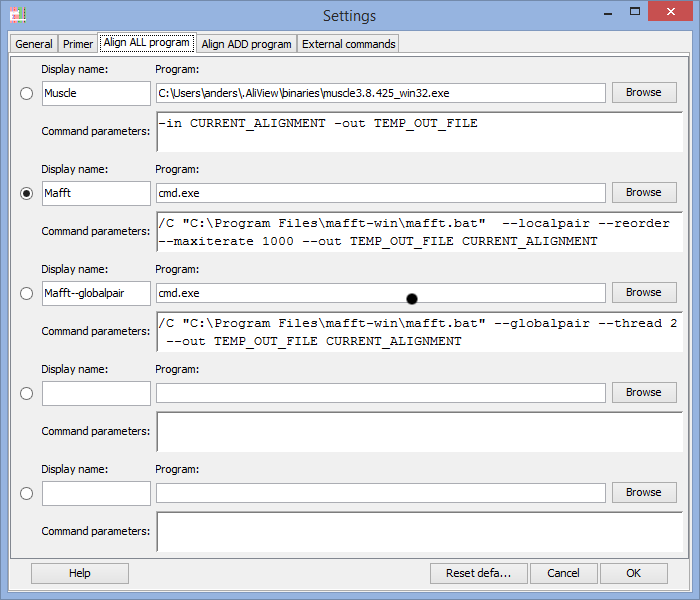
In the Program field you enter the program you want to execute when using the alignment functions in AliView.
There are default settings for MUSCLE and MAFFT included in AliView and they look different for different operating systems (Windows / Linux / Mac OSX).
Below are commented examples for Mac OS X, Linux and Windows
Note 1:It is essential that file names that contains spaces or unorthodox characters are specified within quotation marks "
On Windows for example: "c:\Program Files\mafft-win\mafft.bat"
On Mac OS X for example: "FigTree 1.4.0.App"
Note 2: When calling MAFFT on Windows computers you are actually starting the program cmd.exe and instead including MAFFT program path as a parameter (compare for example with the muscle.exe program and command).
Program to use when adding and aligning in new sequences ("Align add" commands)
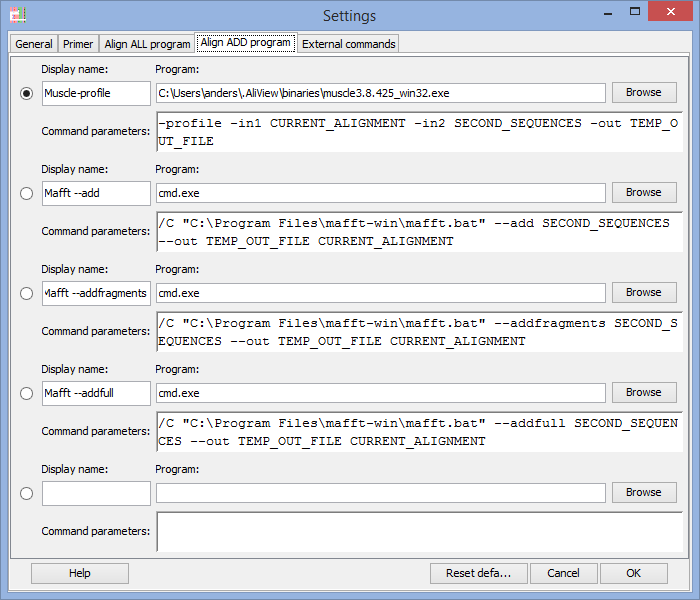
For an explanation on how to fill in the fields see above "Program to use for "Align all" commands" and below are also commented examples for Mac OS X, Linux and Windows
Some commented examples of Aligner Program commands:
NOTE: It is essential that file names that contains spaces are specified within quotation marks "MAC OS X
| Align ALL command examples | |
| MUSCLE command (default) | Comment |
/home/the-user/.AliView/binaries/muscle3.8.425_i86linux64 |
The alignment program (MUSCLE) is run with parameter -in -out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE |
| MAFFT example | Comment |
/usr/local/bin/mafft |
NOTE that MAFFT is not installed with AliView, and
has to be installed separately In this example the program (MAFFT) is run with parameter --localpair --reorder --maxiterate 1000 --out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
| Align ADD command examples | |
| MUSCLE command (default) | Comment |
/home/the-user/.AliView/binaries/muscle3.8.425_i86linux64 |
The alignment program (MUSCLE) is run with parameter -profile -in1
-in2 -out This is the MUSCLE way of adding sequences to an existing alignment. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE |
| MAFFT example | Comment |
/usr/local/bin/mafft |
NOTE that MAFFT is not installed with AliView, and
has to be installed separately In this example the program (MAFFT) is run with parameter --add --out AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
WINDOWS
| Align ALL command examples | |
| MUSCLE command (default) | Comment |
C:\Users\anders\.AliView\binaries\muscle3.8.425_win32.exe |
The alignment program (MUSCLE) is run with parameter -in -out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE |
| MAFFT example | Comment |
cmd.exe |
NOTE that MAFFT is not installed with AliView, and
has to be installed separately In this example the program (MAFFT) is run with parameter --localpair --reorder --maxiterate 1000 --out NOTE2 The program mafft.bat is actually called by program cmd.exe with parameter /C AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
| Align ADD command examples | |
| MUSCLE command (default) | Comment |
C:\Users\anders\.AliView\binaries\muscle3.8.425_win32.exe |
The alignment program (MUSCLE) is run with parameter -profile -in1
-in2 -out This is the MUSCLE way of adding sequences to an existing alignment. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE |
| MAFFT example | Comment |
cmd.exe/C "C:\Program Files\mafft-win\mafft.bat" --add SECOND_SEQUENCES
--out TEMP_OUT_FILE CURRENT_ALIGNMENT_FASTA |
NOTE that MAFFT is not installed with AliView, and
has to be installed separately NOTE2 The program mafft.bat is actually called by program cmd.exe with parameter /C In this example the program (MAFFT) is run with parameter --add --out AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
LINUX
| Align ALL command examples | |
| MUSCLE command (default) | Comment |
/home/the-user/.AliView/binaries/muscle3.8.425_i86linux64 |
The alignment program (MUSCLE) is run with parameter -in -out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE |
| MAFFT example | Comment |
/usr/local/bin/mafft |
NOTE that MAFFT is not installed with AliView, and
has to be installed separately In this example the program (MAFFT) is run with parameter --localpair --reorder --maxiterate 1000 --out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
| Align ADD command examples | |
| MUSCLE command (default) | Comment |
/home/the-user/.AliView/binaries/muscle3.8.425_i86linux64 |
The alignment program (MUSCLE) is run with parameter -profile -in1
-in2 -out This is the MUSCLE way of adding sequences to an existing alignment. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE |
| MAFFT example | Comment |
/usr/local/bin/mafft |
NOTE that MAFFT is not installed with AliView, and
has to be installed separately In this example the program (MAFFT) is run with parameter --add --out AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
External commands - Send the alignment to other programs
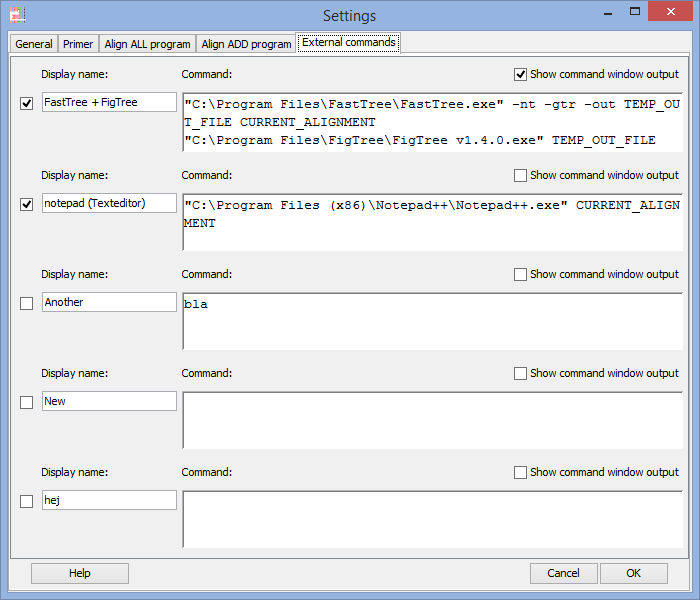
You can send the active alignment from AliView as a command parameter to other programs and create a simple pipeline of commands executed from a Menu-button in AliView.
Each line is executed as a separate command waiting for the previous to have finished. An an example of this would be a simple combination of commands that take the current alignment and run it in FastTree to create a Maximum Likelihood Tree and finally open the resulting tree in FigTree.Special Parameters:
CURRENT_ALIGNMENT_FASTA = The current alignment as a fasta file
CURRENT_ALIGNMENT_PHYLIP = The current alignment as a phylip file
TEMP_OUT_FILE = An empty temporary file created by AliView in the temp-directory of the computer
ALIVIEW_OPEN file-name = Open a new window with file name as parameter
Some examples of external commands with comments:
NOTE: It is essential that file names that contains spaces or unorthodox characters are specified within quotation marks "| MAC OS X | |
| Command | Comment |
/usr/local/bin/FastTree -nt -gtr -out TEMP_OUT_FILE
CURRENT_ALIGNMENT_FASTA |
NOTE that FigTree application name is surrounded with quotation marks
since the name contains a space. NOTE that FigTree application is started with MacOS X command open -a The program FastTree is run with parameters -nt -gtr -out [tree-file-out] [input_alignment] AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment When FastTree.exe is finished creating the tree, it will have saved the result into the file TEMP_OUT_FILE FigTree v1.4.0 is run when FastTree is finished and as parameter to the program is the tree in the file TEMP_OUT_FILE |
| WINDOWS | |
| Command | Comment |
"C:\Program Files\FastTree\FastTree.exe" -nt -gtr -out
TEMP_OUT_FILE CURRENT_ALIGNMENT_FASTA |
NOTE that FastTree.exe and FigTree.exe application
names are surrounded with quotation marks " since the file path and name contains a space. The program FastTree.exe is run with parameters -nt -gtr -out [tree-file-out] [input_alignment] AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment When FastTree.exe is finished creating the tree, it will have saved the result into the file TEMP_OUT_FILE FigTree v1.4.0.exe is run when FastTree.exe is finished and as parameter to the program is the tree in the file TEMP_OUT_FILE |
| LINUX | |
| Command | Comment |
/usr/bin/FastTree -nt -gtr -out TEMP_OUT_FILE
CURRENT_ALIGNMENT_FASTA |
NOTE that the file name should be enclosed in quotation marks if there is space in the path. The program FastTree is run with parameters -nt -gtr -out [tree-file-out] [input_alignment] AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment When FastTree.exe is finished creating the tree, it will have saved the result into the file TEMP_OUT_FILE FigTree v1.4.0 is run when FastTree is finished and as parameter to the program is the tree in the file TEMP_OUT_FILE |
Drag and Drop
If you drop a file in the program window it will open a new window with the
alignment
Note: If you drop a sequence while pressing the "Shift"-button - the
sequences in the file will be added to the currently loaded alignment.
Java 8 JRE Install (Linux)
This is tested on Ubuntu/Debian/OpenSuse (RedHat - just download and install rpm-package)(These installation instructions are compiled and simplified fom this excellent tutorial):
http://install-things.com/2014/03/24/how-to-install-oracle-java-8-on-ubuntu-12-04-linux/
1. Download jre tar.gz-version:
http://www.oracle.com/technetwork/java/javase/downloads/jre8-downloads-2133155.html
2. Unpack
sudo tar zxvf jre-8*.tar.gz
3. Move whole unpacked dir:
sudo mv jre1.8.* /usr/lib/jvm/
4. Tell your system where new java is located:
sudo update-alternatives --install /usr/bin/java java /usr/lib/jvm/jre1.8.*/bin/java 1
5. Tell your system to use new version:
sudo update-alternatives --set java /usr/lib/jvm/jre1.8.*/bin/java
Java 9 JRE Install (Linux)
This is tested on Ubuntu/Debian/OpenSuse (RedHat - just download and install rpm-package)
# 1. Download jre tar.gz-version:
http://www.oracle.com/technetwork/java/javase/downloads/jre9-downloads-3848532.html
# 2. Unpack
sudo tar zxvf jre-9.*.tar.gz
# 3. Make sure jvm-dir exists:
sudo mkdir -p /usr/lib/jvm/
# 4. Move whole unpacked dir:
sudo mv jre-9.* /usr/lib/jvm/
# 5. Tell your system where new java is located:
sudo update-alternatives --install /usr/bin/java java /usr/lib/jvm/jre-9.*/bin/java 1
# 6. Tell your system to use new version:
sudo update-alternatives --set java /usr/lib/jvm/jre-9.*/bin/java